
El Grupo de Coordinación de Productos Médicos (MDCG en inglés) integrado por representantes de todos los Estados Miembro de la Unión Europea ha publicado la Guía en Ciberseguridad para dispositivos médicos en diciembre de 2019.

El motivo de su publicación es que las Regulaciones Medicas para Dispositivos compuestas por la Regulación para Dispositivos Médicos 745/2017 (MDR-https://data.europa.eu/eli/reg/2017/745/2017-05-05) y la Regulación para Dispositivos Médicos In Vitro 746/2017 (IVDR-https://data.europa.eu/eli/reg/2017/746/2017-05-05) que entraron en vigor en mayo de 2017, deben estar totalmente establecidas en mayo de 2020 para el caso del MDR y para mayo de 2022 para el caso de IVDR:
- 05/04/2017 Publicación MDR
- 05/04/2017 Publicación IVDR
- 05/05/2017 Entrada en vigor
- Mayo 2020 MDR totalmente establecida
- Mayo 2022 IVDR totalmente establecida
El principal objetivo de la guía es proveer a los fabricantes de las instrucciones necesarias para cumplir con los requisitos relacionados con la ciberseguridad del Anexo I del MDR e IVDR durante todo el ciclo de vida de los dispositivos. Estos requisitos afectan al hardware, a las características de la red informática y a las medidas de seguridad de TI de forma similar a como sucede en certificaciones de renombre en el ámbito de la ciberseguridad, como son Common Criteria, FIPS 140-2, PCI-PTS o LINCE, las cuales sirven para certificar la conformidad de productos tecnológicos con los requisitos de seguridad que dichos estándares establecen en función del propósito para el que serán utilizados, su funcionalidad y funcionalidades criptográficas que implementa.
La consecuencia más inmediata del establecimiento de ambas regulaciones es que afectarán al a todos los dispositivos médicos que implementen funcionalidades de ciberseguridad que deseen obtener el marcado CE para poder ser comercializados en el marco de la Unión Europea.
Consideraciones generales de seguridad
Las Regulaciones para Dispositivos Médicos consideran que debido a que los dispositivos médicos poseen riesgos asociados a su uso al igual que sucede con cualquier tipo de dispositivo, es necesario poder encontrar el equilibrio adecuado entre los riesgos inherentes a su modo de uso y el mayor nivel de protección de salud y seguridad que puede proveer el dispositivo.
Por un lado, los fabricantes serán los encargados de definir las mejores prácticas de uso asociadas a requisitos de seguridad, de manera que se minimicen o eliminen los posibles riesgos y amenazas de seguridad. Para ello, deberán tener en cuenta el tipo de dispositivo, el entorno de uso y el modo de uso del mismo para definir los controles de seguridad y reglas de uso relacionados con el producto, teniendo en cuenta, además, que en la mayoría de los casos las vulnerabilidades se descubren y se explotan debido a un uso indebido del dispositivo en cuestión.
Por otro lado, los centros médicos donde se hace uso de los dispositivos médicos deberán adecuar sus procesos de gestión de riesgos de manera que se adhieran de la mejor manera posible a las mejores prácticas proporcionando:
- Buen nivel de seguridad física para evitar el acceso no autorizado a los dispositivos médicos o puntos de acceso.
- Medidas de control de acceso basada en roles para acceder a los elementos de red, información almacenada y/o servicios.
- Gestión de parches de seguridad
- Protección contra malware
- Cursos de concienciación para la seguridad
- Métodos para verificar quien ha llevado a cabo cambios en el sistema.
Además de los fabricantes y los centros médicos, es necesario tener en cuenta al resto de actores principales en lo que a los dispositivos médicos se refiere, como son:
- Los integradores: Son los encargados de llevar a cabo la correcta instalación y configuración del dispositivo teniendo en cuenta las instrucciones proporcionadas por los fabricantes. Del mismo modo, darán soporte a los operadores cuando lo requieran.
- Los operadores: Son los responsables de hacer que los dispositivos se usen y mantengan de manera segura según lo indicado por el fabricante.
- Profesionales médicos: Son los encargados de hacer uso del dispositivo médico en si para aplicarlo al paciente el tratamiento requerido. Por tanto, deberán ser instruidos para hacer un uso adecuado del dispositivo desde el punto de vista de la ciberseguridad.
- Pacientes: Son los usuarios que recibirán el tratamiento adecuado mediante el dispositivo médico y al igual que los profesionales médicos, deberán ser instruidos para hacer un uso adecuado del dispositivo desde el punto de vista de la ciberseguridad en caso de que puedan consultar sus datos médicos o tengan algún tipo de interacción con el dispositivo.
Diseño y fabricacion de seguros
Al igual que sucede con cualquier dispositivo IT, la seguridad, la protección y la eficiencia deben ser aspectos a tener en cuenta durante el todo el ciclo de vida del dispositivo, englobando así desde los procesos de diseño y fabricación hasta el proceso de decomisado
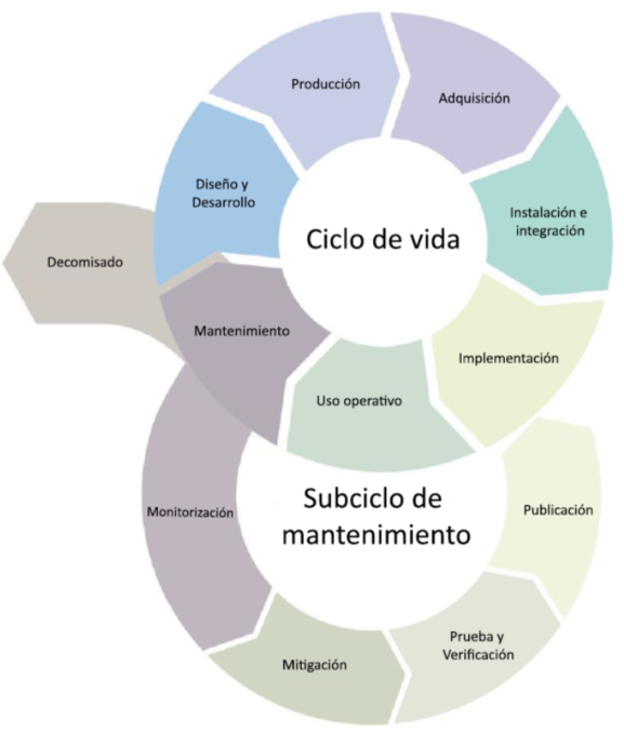
Para conseguir un nivel de seguridad adecuado es necesario que la misma se tenga en cuenta desde la etapa de diseño, ya que, en caso contrario, puede suceder que, en etapas avanzadas del proyecto, sea necesario volver atrás para redefinir requisitos de fabricación que satisfagan los requisitos de seguridad necesarios.
Para ello, la guía plantea que se realice durante todo el ciclo de vida del producto llevando a cabo la siguiente lista de buenas prácticas:
- Administración de la seguridad: Es necesario planificar y documentar todas las actividades de seguridad necesaria para cumplir con los requisitos de seguridad establecidos en el Anexo I de los documentos MDR e IVDR. Es decir, tras analizar dichos requisitos de seguridad, se debe establecer un plan de gestión de la seguridad para asegurar que los requisitos se cumplen durante todo el ciclo de vida del dispositivo.
- Determinar los requisitos de seguridad necesarios: Consiste en identificar las funciones de seguridad necesarias a cumplir por el dispositivo inherentes a los requisitos al Anexo citado en el paso anterior para asegurar la confidencialidad, integridad y disponibilidad de los datos según su propósito.
Esto implica determinar si es necesario implementar medidas de seguridad físicas o que se lleve a cabo la protección de los interfaces externos. Este tipo de protección, es muy común en certificaciones como FIPS 140-2 o Common Criteria, donde es común hacer uso de medidas anti tampering basadas en sellos que evidencien los intentos de manipulación del dispositivo o el uso de resinas epoxy y circuitos de específicos que eviten el acceso al hardware del dispositivo o que lo dejen inoperativo en caso de intentarlo.
Por otro lado, también implica el determinar si es necesario llevar cabo a la implementación de mecanismos de autenticación, autorización, cifrado o auditoría. Al igual que para los requisitos de seguridad físicos, estas medidas de seguridad a nivel de software nos recuerdan a requisitos de seguridad utilizados en FIPS 140-2 donde se requiere hacer uso de medidas de autenticación basadas en roles así como los auto test de para verificar la integridad del software comprobando que no ha sido modificado, o a los requisitos de seguridad utilizados en Common Criteria donde ser requiere hacer uso de logs de auditoría para verificar quien ha tenido acceso a funcionalidades de seguridad de dispositivo.
- Verificación de la seguridad y test de validación: Esta etapa requiere definir los test necesarios para verificar que el producto cumple con todos los requisitos de seguridad durante su ciclo de vida total. De nuevo, esta práctica es similar a la realizada en la fase ATE_FUN en las certificaciones de Common Criteria o a la que se requiere en FIPS 140-2 donde es necesario llevar a cabo un conjunto de auto test para verificar la integridad del software, así como la correcta operatividad del dispositivo cada vez que este se enciende o cada vez va a usar una funcionalidad específica.
- Gestión de incidencias de seguridad: Este proceso se debe llevar a cabo para determinar cómo se deben gestionar las posibles incidencias de seguridad. Una forma correcta de llevar a cabo este proceso podría ser el planteado por la familia ALC_FLR de la certificación Common Criteria, el cual establece que el fabricante debe mantener un tracking de las vulnerabilidades descubiertas y de cómo remediarlas durante todo el ciclo de vida del dispositivo. Otro ejemplo puede ser, hacer que el dispositivo se bloquee requiriendo asistencia técnica en caso de que se detecte una incidencia de seguridad para evitar que los pacientes resulten dañados debido a un comportamiento inadecuado del dispositivo.
- Gestión de actualizaciones de seguridad: El proceso se encarga, por un lado, de determinar se testearán las actualizaciones de seguridad de modo que el equipo pueda verificar que es legítima por medio de la verificación de su hash, y, por otro lado, de definir el periodo de tiempo y la forma en la que se hará que estén disponibles.
- Normas de seguridad: Este proceso determina la documentación de usuario que describe como llevar a cabo la integración, configuración y mantenimiento del producto para operar de forma segura. De nuevo este apartado nos recuerda a las guías de instalación (AGD_PRE) y de uso (AGD_OPE) de Common Criteria y FIPS 140-2.
A continuación, se detallan las fases necesarias para implementar la lista de buenas prácticas detallada anteriormente, para así considerar la ciberseguridad desde el inicio evitando tener que volver atrás en etapas avanzadas del desarrollo:
Gestión de los riesgos de la seguridad
Este proceso consiste en llevar a cabo un análisis y control de riesgos especificando las medidas a llevar a cabo para mitigarlos en caso de que ocurran hasta un nivel aceptable (ver Anexo I sección 17.1 (MDR) y sección 16.1 (IVDR)). Este proceso es una parte vital en otras certificaciones como en Common Criteria, donde la familia ASE_SPD determina la forma correcta a seguir por el fabricante para documentar todas las posibles amenazas del sistema.
Las funciones de seguridad que hacen que el dispositivo cumpla con los requisitos de seguridad del Anexo I de los documentos MDR e IVDR se deben establecer como medidas de control de riesgos para eliminar o reducir los riesgos detectados en el análisis de riesgos, estableciendo sistemas de alarmas o avisos para los riesgos que no se puedan eliminar y proporcionando información acerca de los avisos que se generan. Esta guía proporciona la siguiente lista de posibles funciones de seguridad a cumplir por el dispositivo, la cual es similar a los requisitos a cumplir por un dispositivo en una evaluación LINCE o a las funciones de seguridad (SFRs) a cumplir por un dispositivo en una evaluación Common Criteria:
- Cierre automatico de sesión
- Controles de auditoría
- Autorización
- Configuración de las herramientas de seguridad
- Desidentificación de los datos personales
- Copias de seguridad y recuperación de desastres
- Acceso de emergencia
- Integridad y autenticidad de los datos personales
- Detección/Protección de malware
- Autenticación de nodos
- Autenticación de personas
- Bloqueos físicos
- Hardenización del sistema y SO
- Guias de seguridad y privacidad
- Confidencialidad del almacenamiento de datos personales
- Confidencialidad de la transmisión
- Integridad de la transmisión
Evaluación de riesgos de seguridad
Después de elegir las funcionalidades de seguridad, el siguiente paso es el de determinar cual es el entorno de ejecución más apropiado para encontrar el mejor equilibrio entre seguridad, protección y efectividad, para ello hay que llevar a cabo una identificación de los posibles vectores de ataque, así como de las vulnerabilidades asociados a los mismos, usando para ellos sistemas de clasificación de vulnerabilidades como el CVE para asignarles prioridad a cada una de ellas.
Posiblemente la forma más adecuada de que el fabricante lleve a cabo la evaluación de riesgos de seguridad e identifique lo posibles vectores de ataque, es la recomendada por la familia AVA_VAN de la certificación Common Criteria.
Análisis de aceptación de riesgos de seguridad
Se basa en establecer los criterios de aceptación de riesgos de seguridad en función de propósito del dispositivo y su entorno de operación.
Requisitos TI mínimos
El fabricante debe proporcionar documentación acerca del uso, configuración de seguridad y controles de seguridad relacionados con el entorno de operación (conforme a las secciones 17.4 de MDR y 16.4 de IVDR) y tenerla disponible y actualizada de forma electrónica para los usuarios (conforme a secciones 23.4 MDR y 20 IVDR).
Esta documentación es similar a la requerida por certificaciones como Common Criteria, FIPS 140-2 y LINCE las cuales requieren especificar las condiciones del entorno y como proceder para llevar a cabo la configuración segura del dispositivo.
Verificación/validación
La principal medida de seguridad a usar es el testing, basado en llevar a cabo fuzz testing, escaneo de vulnerabilidades, test de penetración y análisis de código. Estas medidas de validación son similares a las requeridas por el estándar PCI-PTS, el cual lleva a cabo pruebas de penetración sobre el propio dispositivo y requiere evidencias de haber llevado a cabo fuzz testing y análisis de código al fabricante.
Aspectos del ciclo de vida
Aunque abordar los riesgos de seguridad desde el diseño ayuda a mitigar los posibles riesgos y amenazas, es necesario llevar a cabo tareas de mantenimiento relacionadas con la seguridad, debido a que en el momento de su certificación un dispositivo medico puede considerarse seguro conforme a las vulnerabilidades conocidas en ese momento, sin embargo, pueden surgir nuevos vectores de ataque que hagan que deje de ser seguro, por tanto, es necesario que durante todo el ciclo de vida, los fabricantes realicen un registro de la información de detallada a continuación:
- Incidentes relacionados con el software del dispositivo.
- Vulnerabilidades de seguridad relacionadas con el hardware del dispositivo y con el hardware de terceros integrado en el mismo.
- Existencia de nuevos vectores de ataque.
El fabricante deberá evaluar los riesgos detectados y llevar a cabo la actualización del software o la modificación del hardware necesaria en las instalaciones del operador.
Documentación e instrucciones de uso
La documentación que el fabricante debe proporcionar al organismo notificado para ser evaluada, debe contener la información que demuestre el cumplimiento de los requisitos generales de seguridad y comportamiento especificados en el Anexo I de las regulaciones para Dispositivos Médicos. Esto significa que el fabricante deberá generar un documento similar a la Security Target de Common Criteria y LINCE o la Security Policy de FIPS 140-2 y PCI-PTS y que en caso de no hacerlo o no cumplir con lo requerido, no podrá obtener el certificado que lo habilite a la venta de su dispositivo en el marco de la Unión Europea.
Además, esta documentación deberá mantenerse actualizada con la información relativa a las vulnerabilidades descubiertas tras la venta del dispositivo y como remediarlas.
Relación con otras legislaciones
Estas Regulaciones para Dispositivos Médicos (MDR e IVDR) deben de aplicarse en paralelo a lo especificado por la Directiva NIS que establece las medidas legales para impulsar el nivel de la ciberseguridad en el marco de la UE y el RGPD (Reglamento General de Protección de Datos) que se encarga de regular el procesamiento de datos personales de los individuos por parte de las empresas en la UE.
Por un lado, el objetivo de la Directiva NIS es el de asegurar que los estados miembro de la UE cuenten con un Equipo de Respuesta a Incidentes de Seguridad y una autoridad NIS competente, de manera que puedan cooperar entre sí de manera eficiente mediante el intercambio de información para dar respuesta a los incidentes de ciberseguridad y favoreciendo la existencia de una cultura en cuanto a seguridad de los servicios vitales e infraestructuras críticas para la sociedad que dependen de las TIC como son la energía, transporte, agua, banca, etc.
Por otro lado, el objetivo del RGPD es el de tratar de asegurar la protección de los datos personales de manera que no sea posible asociar datos o partes de ellos con el usuario al que pertenecen usando para ello técnicas de cifrado y pseudoanonimización. Esto es necesario debido a que como establece el propio RGPD, los datos médicos de un usuario son considerados datos personales de alto riesgo.
Conclusión
Esta Guía en Ciberseguridad para la certificación de dispositivos Médicos por parte de los organismos notificados designados por cada país, ofrece una visión general de los principales aspectos a considerar por los fabricantes de cara a cumplir con los requisitos de seguridad establecidos por el Anexo I del MDR e IVDR, facilitando enfrentarse a la tarea de generar la documentación necesaria para para cumplir con ambos reglamentos en función del tipo de dispositivo médico y habilitando así, a los dispositivos que consigan obtener el certificado de la UE (marcado CE) a que puedan ser introducidos en el mercado de la Unión Europea portando la etiqueta de marcado CE junto con el número de identificación del organismo notificado que ha llevado a cabo la evaluación del producto.
Así mismo, la existencia de ambos reglamentos (MDR e IVDR) evidencia el hecho de que no es válido un esquema basado en la auto evaluación, donde el fabricante sea el encargado de validar su producto, sino, que se necesita un esquema donde un laboratorio (organismo notificado) sea el encargado de verificar que la documentación cumple con lo estipulado en ambos reglamentos y al mismo tiempo el dispositivo cumple con lo especificado en la documentación, reduciendo así la cantidad de dispositivos médicos con fallos de ciberseguridad que acaban afectando a los pacientes y a su privacidad.










