
The Medical Device Coordination Group (MDCG) composed of representatives of all European Union Member States has released the Guidance on Cybersecurity for medical devices on December 2019.

The main reason for its publication is that the Medical device Regulation 745/2017 (MDR) and the In Vitro Medical Device Regulation 746/2017 (IVDR) released on May 2017 must be totally stablished on May 2020 in case of MDR and on May 2022 in case of IVDR:
- 05/04/2017 MDR Publication
- 05/04/2017 IVDR Publication
- 05/05/2017 Entry into force
- Mayo 2020 MDR totally established
- Mayo 2022 IVDR totally established
The aim of this guidance is to provide the manufacturers with the needed instructions to comply with the requirements related to the cybersecurity contained in Annex I of the MDR and IVDR for the entire life cycle of the devices. These requirements affect the hardware, IT networks characteristics and IT security Measures in a similar form as occurs in well-known cybersecurity certifications, such as Common Criteria, FIPS 140-2, PCI-PTS or LINCE, which certifies the conformity of the technological products with the security requirements established by them with regards to the purpose of the products, their features and cryptographic functionalities
The most immediate consequence is that both regulations (MDR e IVDR) will impact all the medical devices to be evaluated to obtain the CE marking that implement cybersecurity functionalities.
General security considerations
The Medical Devices Regulations consider that the medical devices have risks associated with their use as any type of device, therefore it is necessary to find the adequate balance between the risks related to their mode of use and the higher level of health protection and security that the device is able to provide.
Firstly, the manufacturers are responsible for defining the best practices of use associated with the security requirements in order to minimize or to eliminate the possible security risks and threads. To do that, they shall consider the type of device, its environment and its mode of use to define the security controls and rules of use related to the product. In addition, it will be necessary to consider that in most cases, the vulnerabilities usually are discovered because of the improper use of the device.
Secondly, the medical centres where the devices will be used shall adequate their risks management processes to get a better adequation to the best practices in order to provide:
- A good level of physical security to avoid non-authorised access to medical devices or access points.
- Role-based access control measures to access to the network elements, stored information and/o services.
- Security patches management.
- Malware protection
- Cybersecurity awareness courses.
- Methods to verify who has implemented the changes to the system.
In addition to the manufacturers and medical centres, it is necessary to take into account the other main actors regarding medical devices, such as:
- The integrators:They are in charge of carrying out the correct installation and configuration of the device taking into account the instructions provided by the manufacturers. Moreover, they will give support to the operators if they require it.
- The operators:They are responsible for assuring that the devices are used and maintained as detailed by the manufacturer.
- Medical professionals:They use the device to apply the required treatment to the patients. Therefore, they must be instructed in order to use the device in the correct way with regard to the cybersecurity.
- Patients:They will receive the treatment provided by the medical device and in the same way as the medical professionals, they must be instructed to use the device in an adequate way with regard to the cybersecurity if exist the possibility of they can interact with the device.
Secure design and production
In the same way, as for any TI device, the security, the safety and efficiency must be considered for the entire life cycle of the device encompassing from the design process to decommission process.
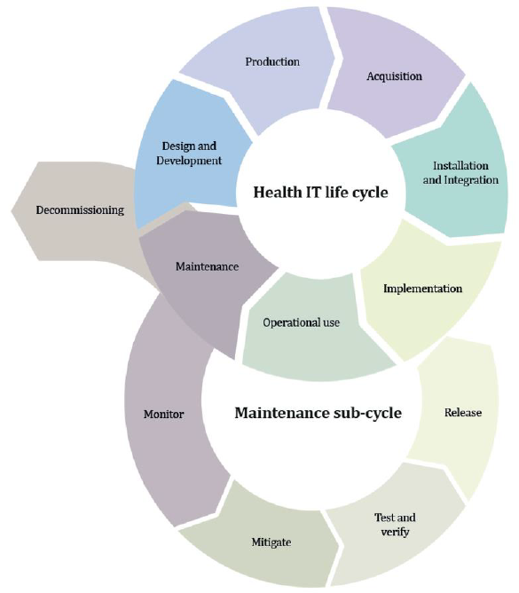
In order to get a good level of security it is necessary to consider it from the design stage, because in otherwise, it may imply to go back in advanced stages to redefine manufacturing requirements that comply with the needed security requirements
To do that, the guidance states that security must be considered during the entire product life cycle by performing the following list of good practices:
- Security management:It is necessary to plan and document all the activities related to the security to comply with the security requirements detailed in Annex I of the MDR and IVDR documents. Hence, a security management plan must be defined to ensure that the security requirements are satisfied during the entire device life-cycle.
- Security requirement identification:It consists of identifying the required security functions to comply with the security requirements defined in Annex I of the MDR in order to ensure the confidentiality, integrity and availability of the data depending on its purpose.
This implies to determine if it is necessary to implement physical security measures or to protect the external interfaces. This type of protection is usually used in certifications like FIPS 140-2 or Common Criteria, which usually require to use antitamper measures based on tamper-evident seals or epoxy resins and specific circuits that evidence the tamper attempts to access to the hardware or that make it inoperative.
On the other hand, it also involves determining if it is necessary to implement authentication, authorization, encryption or auditing mechanisms. As occur with the physical requirements, this software measures are similar to security requirements of FIPS 140-2 which requires to use role-based authentication measures and self-tests to verify the integrity of the software checking that it has not been verified, or to the security requirements of Common Criteria that require to use audit logs to verify who has access to the security functions of the device.
- Security verification and validation test:This stage requires to define the necessary test to verify that the product complies with all the security requirements during its total life cycle. Again, this practice is similar to the carried out in the ATE_FUN stage in the Common Criteria certifications or to the required by FIPS 140-2 which requires to carry out a set of self-test to verify the software integrity, as well as the correct operativity of the device when it is powered on or it is going to use a specific functionality.
- Security verification and validation test: This stage requires to define the necessary test to verify that the product complies with all the security requirements during its total life cycle. Again, this practice is similar to the carried out in the ATE_FUN stage in the Common Criteria certifications or to the required by FIPS 140-2 which requires to carry out a set of self-test to verify the software integrity, as well as the correct operativity of the device when it is powered on or it is going to use a specific functionality
- Security incidents management:This process is carried out to determine how to manage possible security incidents. A correct way to perform this process is the specified by the ALC_FLR family of the Common Criteria certification that establishes that the manufacturer must maintain tracking of the discovered vulnerabilities and how to mitigate them during all the device life cycle. Another example can be that the device remains locked requiring technical assistance in case it detects a security incidence to avoid that the patients result damaged because of the bad behaviour of the device.
- Security rules:This process specifies the user documentation that defines how to carry out the integration, configuration and maintenance of the product to operate in a secure mode. Again, this apart remembers us to the installation guidance (AGD_PRE) and use (AGD_OPE) of Common Criteria and FIPS 140-2.
Here below are detailed the phases to implement the best practice list specified above, in order to considerate the cybersecurity from the beginning avoiding to have to go back in advanced stages of the device development.
Security risk management
This process consists of performing a risk control and analysis detailing the measures to mitigate them if they occur to an acceptable level (see Annex I - section17.1 (MDR) and section 16.1 (IVDR)). This process is one of the main parts in other certifications such as Common Criteria, where the ASE_SPD family determines the correct way to be followed by the manufacturer to document all the possible system threats
Security Functions
The security functions that make the device to comply with the security requirements of the Annex I of the MDR and IVDR must be established as risk control measures to remove or reduce the risks detected in the risk analysis, establishing alarms or warning systems to the risks that can not be removed, providing information about the generated warnings. This guidance provides the following list of possible security functions to be complied by the device, which is similar to the security requirements to be complied in a LINCE or Common Criteria evaluation:
- Automatic Logoff
- Audit Controls
- Authorization
- Configuration of Security Features
- Cybersecurity Product Upgrades
- Personal Data De-Identification
- Data Backup and Disaster Recovery
- Emergency Access
- Personal Data Integrity and Authenticity
- Malware Detection/Protection
- Node Authentications
- Person Authentication
- Physical Locks
- System and OS hardening
- Security and Privacy Guides
- Personal Data Storage Confidentiality
- Transmission Confidentiality
- Transmission Integrity
Security risk evaluation
After choosing the security functionalities, the following step is to determine what is the execution environment more appropriated to find the best balance between security, safety and effectiveness, for that it is necessary to carry out an identification of the possible attack vectors and the vulnerabilities associated to each of them using the classification systems of vulnerabilities as the CVE to assign a priority for each of them.
Possibly, the best way that can be used by the manufacturer to identify the possible attack vectors is the recommended by the AVA_VAN family of the Common Criteria certification
Security Benefit-Risk analysis
It is based on establishing the security risk acceptance criteria depending on the device purpose and its execution environment
Minimum IT requirements
The manufacturer must provide documentation about the use, security configuration and security controls related to the operating environment (in accordance with sections 17.4 of the MDR and 16.4 of the IVDR) and maintain it available and updated for the users (as detailed in sections 23.4 of the MDR and 20 of the IVDR).
This documentation is similar to the required by other certifications like Common Criteria, FIPS 140-2 and LINCE which require to specify the environmental conditions and how to proceed to carry out the secure configuration of the device.
Verificación/validación
The main security measure is testing. It consists of executing fuzz testing, vulnerability scans, penetration testing and code analysis. These validation measures are similar to the required by the PCI-PTS standard which performs penetration testing to the device and requires evidence of that the fuzz testing and code analysis have been executed.
Life cycle aspects
Although addressing security risk from design can help to mitigate the possible risks and threats, it is necessary to implement maintenance tasks related to the security, because it is possible that the device can be considered secure in the moment of the certification but, however, new vector attacks can appear later and make the device insecure, therefore, it is necessary that the manufacturers record the following information during the entire device life cycle:
- Security incidents related to the device software.
- Security vulnerabilities related to the device hardware and to the third-party hardware integrated with it.
- Existence of new attack vectors.
The manufacturer shall evaluate the detected risks and implement the software updates or hardware modifications required to mitigate them at the operator facilitates.
Documentation and instructions for use
The documentation provided by the manufacturers to the notified organisms to be evaluated, must contain the information that shows the compliance with the general security and behaviour requirements specified in Annex I of the Medical Device Regulations. This means that the manufacturer shall generate a document similar to the Security Target of the Common Criteria and LINCE evaluations or to the Security Policy of FIPs 140-2 and PCI-PTS evaluations. If the manufacturer does not generate the documentation or does not comply with the required by the norm, then its device will not obtain the certificate that enables it to be sold in the European Union.
Moreover, this documentation shall be updated with the information related to the vulnerabilities discovered and how to solve them after the device sale.
Relation with other legislations
This Medical Devices Regulations (MDR and IVDR) must be applied in conjunction with the specified by the NIS directive that establishes the legal measures to increase the level of the cybersecurity in the UE framework and the GDPR ( General Data Protection Regulation) that is responsible for regulating the personal data processing of the users by the companies in the European Union.
On one hand, the objective of the NIS directive is to ensure that the member states of the UE are appropriately equipped with a Computer Security Incident Response Team (CSIRT) and a competent national NIS authority, so that they can cooperate efficiently together interchanging information to give response to the cybersecurity incidents and encouraging the existence of a culture of security which is vital for the critical infrastructures that depend on the ICT as the energy, transport, water, banking, etc.
On the second hand, the aim of the GPDR is to ensure the personal data protection in order to avoid the association of data or a part of them with the user to whom they belong using encryption and pseudoanonimization techniques. This is necessary because as it is specified by the GPDR, the medical data of a user are considered as high-risk personal data.
Conclusion
This Cybersecurity Guidance for the Medical Devices certification performed by the notified organisms designated by each country offers a general vision of the main aspects to be considered by the manufacturers in order to comply with the security requirements established on the Annex I of the MDR and IVDR, facilitating the task of generating the necessary documentation to comply with both regulations depending on the type of medical device and enabling the devices that obtain the UE certificate (CE marking) to be introduced in the market of the UE with the making CE label containing the identification number of the notified organism that has performed the product evaluation.
In addition, the existence of both regulations (MDR and IVDR) evidences the fact that a scheme based on self-assessment, where the manufacturer is responsible for validating its product, is not valid. Instead, it is needed a scheme where a laboratory (notified body) is in charge of verifying that the documentation complies with both regulations and at the same time the device complies with what is specified in the documentation, thus reducing the number of medical devices with cybersecurity failures that end up affecting patients and their privacy.










